Designing lubricants by artificial intelligence—the sequel
Drs. Wilfred T. Tysoe & Nicholas D. Spencer | TLT Cutting Edge August 2020
New high-viscosity index oils have been designed by combining autonomous searching with ultrafast molecular-dynamics algorithms.
To be brutally honest, the main tools for developing new lubricants over the last century have been trial and error, combined with luck and a good nose for promising directions. Examples of the latter include the observation that the antioxidant ZnDTP was reducing wear, later leading to its incorporation as an antiwear additive into virtually every motor oil worldwide.¹ Computers, modeling and artificial intelligence (AI) have long shown promise for helping us in this endeavor, but so far their impact on finding new lubricants has been limited.
In the last column,² we described how AI can be used to design and select 2D materials as solid lubricants. The approach used machine learning to uncover critical parameters from a number of known lubricious 2D materials and to predict further materials of the same class that might present improved properties. While this in itself is an impressive achievement, what if we were not limited to the class of materials that we already know and truly could investigate uncharted territory?
Seiji Kajita, Tomoyuki Kinjo and Tomoki Nishi of the Toyota Central R&D Labs in Aichi, Japan, have done exactly that. In a recent paper published in the journal Communications Physics (not necessarily an obvious place for tribologists to browse),³ they describe how they carried out a high-throughput, autonomous search for new organic molecules with promising viscosity-index (VI) properties. They evaluated tens of thousands of molecules by molecular-dynamics (MD) simulations, narrowing them down to a few promising candidates that also showed other practically useful properties. To put the cherry on the top of this extraordinary cake, they then synthesized one of these and measured its VI, which proved to be very high.
The autonomous search scheme used in this work is a so-called “closed-loop search,” in which molecular structures are generated within certain limits of structure, elemental composition and molecular size by a technique known as Monte Carlo Tree Search (MCTS), and they are then evaluated by MD simulations. The results are fed back into the MCTS, and the evaluated molecular structures become continuously optimized for the desired property. A problem here is that MD calculations of viscous properties are notoriously slow. This is partially because viscosity, as a transport property, normally requires long time averages to be taken over large systems, which necessitates a huge number of floating-point operations.
Kajita and colleagues have successfully removed this bottleneck by using an elastic concept of liquid viscosity, originally developed to analyze glass transitions, and known as the “shoving model” (
see Figure 1). This relies on the idea that any molecule in a liquid is surrounded by a “cage” of other molecules, each colliding with each other due to thermal fluctuations. After a relaxation time, the molecule escapes from the cage by pushing away the other molecules, and the combination of many such processes corresponds to macroscopic flow. The energy barrier for this process corresponds to the shear modulus of the liquid, which can be incorporated into an Arrhenius-type equation of viscosity. This approach enables the viscosity and VI to be calculated at least three orders of magnitude more rapidly than by traditional methods.
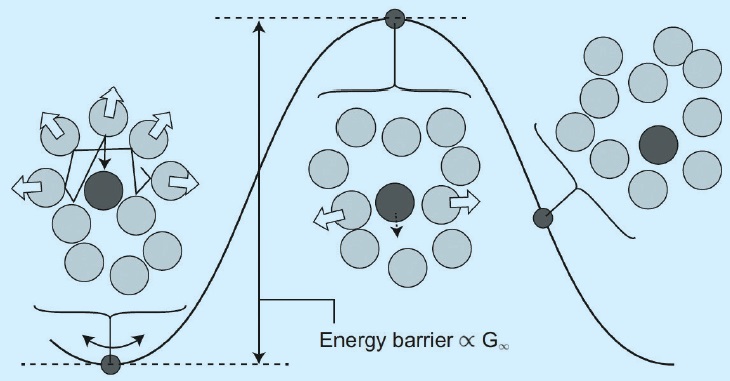 Figure 1. A flow event in the shoving model. The label G∞ indicates shear modulus of the liquid.³
Figure 1. A flow event in the shoving model. The label G∞ indicates shear modulus of the liquid.³
The search algorithm was provided with potential molecular fragments and was instructed to avoid unbranched molecules (which typically have excessively high pour points) and maintain a molecular size corresponding to a reasonable viscosity. The resulting closed-loop search evaluated over 54,000 molecular structures within a viscosity range of 3-6 mm²/s. Many of these had interesting VI properties. In order to test the approach, one of the best-evaluated molecules was identified (
see Figure 2a), and a similar but easier-to-synthesize version (
see Figure 2b) was actually produced and evaluated. The VI properties were either similar to or better than two tested commercial, high-VI base oils, depending on the mode of testing, despite the very non-traditional structure and composition of the molecule.
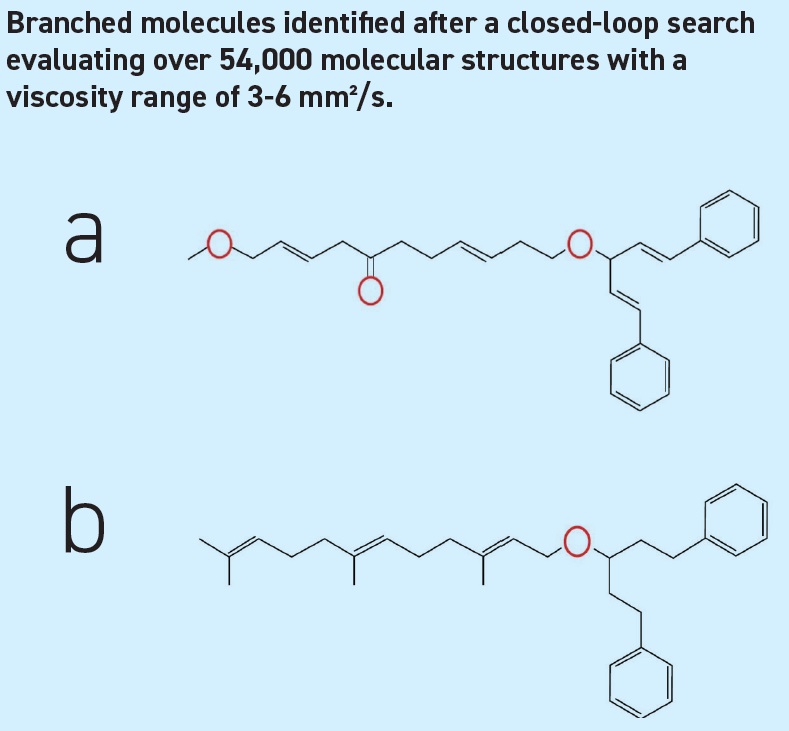 Figure 2. (a) Selected molecule from those showing promise after the autonomous search; (b) similar molecule that could be readily synthesized and which was subsequently evaluated.³
Figure 2. (a) Selected molecule from those showing promise after the autonomous search; (b) similar molecule that could be readily synthesized and which was subsequently evaluated.³
Apart from the elegance and originality of this approach to designing new lubricants, this is an extraordinarily significant development. It shows that simulations, when suitably guided by results from fundamental, experimental and theoretical studies, can be combined with AI, pushing the envelope to come up with some truly new directions in lubricant design. We look forward to further exciting results in this area.
FOR FURTHER READING
1.
Spikes, H. (2004), “The history and mechanisms of ZDDP,”
Tribology Letters,
17, pp. 469-489.
2.
Tysoe, W.T. and Spencer, N.D. (2020), “Designing lubricants by artificial intelligence,” TLT,
76 (6), p. 78. Available
here.
3.
Kajita, S., Kinjo, T. and Nishi, T. (2020), “Autonomous molecular design by Monte-Carlo tree search and rapid evaluations using molecular dynamics simulations,”
Communications Physics,
3, pp. 1-11.
Eddy Tysoe is a distinguished professor of physical chemistry at the University of Wisconsin-Milwaukee. You can reach him at wtt@uwm.edu.
Nic Spencer is professor of surface science and technology at the ETH Zurich, Switzerland, and editor-in-chief of STLE-affiliated Tribology Letters journal. You can reach him at nspencer@ethz.ch.